Overview
Senior Investigator

Research Areas (IRP Lab Groups)
Cytokines are critical for host defense but are also key factors in immune and inflammatory diseases. Innate and adaptive lymphocytes, including T cells, are important selective producers of cytokines, and it is through the production of these that immune responses work together to eliminate microbial pathogens. Host defense against pathogenic microorganisms requires this elegant means of communication between innate and adaptive arms of the immune system. Indeed, cytokines produced by various types of lymphocytes exquisitely tailor immune responses that effectively eliminate intracellular and extracellular pathogens when regulated properly. On the other hand, hyper-active lymphocytes can drive allergic and autoimmune diseases. Conversely, loss-of-function mutations of lymphocyte genes can cause primary immunodeficiencies.
The goal of the Molecular Immunology and Inflammation Branch (MIIB) is to understand how lymphocytes differentiate to selectively produce key immunoregulatory cytokines and to better define the molecular basis of cytokine action.
Discovery of JAK3 and Development of Jakinibs
We now know that a plethora of cytokines is produced by innate immune cells and that these cytokines are critical for the development and specification of CD4 subsets. A substantial portion of cytokines (more than 60) binds to receptors that associate with a class of protein tyrosine kinases termed Janus Kinases or JAKs (illustrated in Figure 2). The MIIB first cloned human JAK3, a kinase predominantly expressed in immune and hematopoietic cells; in partnership with NIH colleagues, the branch went on to demonstrate that JAK3 associates with the common γ chain (γc). This shared cytokine receptor is used by interleukin-2 (IL-2), IL-4, IL-7, IL-9, IL-15, and IL-21, and we found that mutations of JAK3 underlie autosomal recessive severe combined immunodeficiency (SCID). Because of the critical functions of JAKs in cytokine signaling, pharmacological JAK inhibitors (jakinibs) were developed as a new class of immunomodulatory drugs. The NIH holds patents pertaining to Janus Kinases and identification of immune modulators (United States patents 7,070,972 and 7,488,808). The NIAMS partnered with Pfizer in a Cooperative Research and Development Agreement (CRADA), leading to the development of multiple jakinibs. At present, there are nine approved jakinibs for indications ranging from rheumatoid arthritis, psoriatic arthritis, juvenile arthritis, ulcerative colitis, atopic dermatitis, graft-vs-host disease, and myeloproliferative neoplasms to COVID-19. The MIIB, in collaboration with other investigators, is studying the efficacy of jakinibs in lupus, Sjogren’s syndrome, and other disorders.

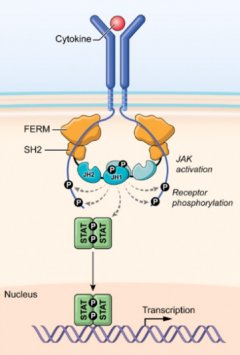
Cytokine Signaling via STATs
STAT4
The MIIB demonstrated that IL-12 activates STAT4, a key factor that initiates the development of Th1 cells that produce the signature cytokine interferon-γ (IFN-γ). The branch has continued its interest in dissecting the function of STAT4 over the last 16 years since this initial discovery. Most recently, we have used second-generation, deep sequencing technology to define the genome-wide targets of STAT4 and relate this knowledge to the roles of STAT4 in regulating gene expression and changes in the epigenome of differentiating CD4+ T cells. Capitalizing on the opportunities provided by deep sequencing technology and computational biology tools, defining the genome-wide functions of STAT4 and other STATs is a focal point of research in the lab.
STAT3
Th17 cell differentiation is dependent upon this transcription factor. Importantly, the Job's syndrome or Hyper IgE syndrome (HIES) disorder is due to mutations of STAT3. The MIIB, in collaboration with other NIH scientists, showed that a major defect in this disorder is impairment of Th17 cell generation. We used Chip-seq methodology to identify STAT3 target genes in developing Th17 cells. Dissecting the functions of STAT3 in this intriguing subset of cells is an ongoing interest of the lab.

STAT1
Patients with STAT1 gain-of-function mutations have clinical presentations similar in many respects to patients with HIES. We are currently studying mouse models to increase our understanding of the pathophysiology of the rare Mendelian disease, which may provide insights into more common diseases.
STAT5
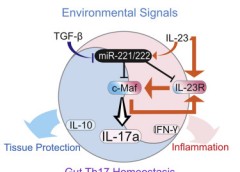
In addition to Th cells, regulatory T (Treg) cells represent another important immunoregulatory subset of CD4+ T cells. The MIIB showed that STAT5 is critical for Treg cell differentiation, binding to the gene encoding the transcription factor Foxp3. Conversely, we showed that STAT5, in response to IL-2 stimulation, also inhibits Th17 differentiation. We further showed STAT5 and STAT3 compete in the Il7a-Il17f locus and that this is an important mechanism underlying the ability of IL-2 to limit Th17 differentiation.
Latest Research into Transcriptome and Epigenome
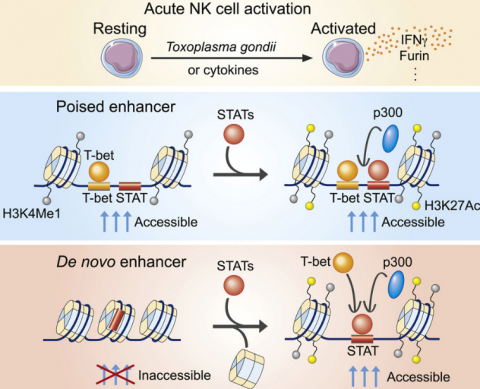
A major ongoing issue in lymphocyte biology is the extent to which different subsets behave as terminally differentiated lineages or retain flexibility in their differentiation programs. By assessing the epigenomes of the different helper cell subsets, we have identified mechanisms through which flexibility is retained, even in polarized "lineages." We have explored the issue of plasticity and heterogeneity of helper T cells and their relationship to innate lymphocytes. We have also explored in detail the structure of key cytokine loci including the Ifng/Il22 and Il4/Il13/Il5 super-enhancer loci.
Furthermore, we study how long noncoding RNAs and microRNAs regulate immune responses, how activation globally impacts chromatin accessibility and architecture, and how these concepts relate to lymphocyte function.
Future Directions
By studying how the engagement of cytokine receptors transduces signals that, in turn, regulate transcription factors and epigenetic events to modulate gene expression, we hope to understand in detail how lymphocytes participate in host defense and contribute to the pathogenesis of immune-mediated diseases. The insights gained from these studies facilitate the development of new therapeutic approaches.
Related NIAMS Labs
Core Research Facilities
Labs at the NIAMS are supported by the following state-of-the-art facilities and services:
Staff

Former Lab Members
U.S. Based
Assistant Professor, Dept. of Internal Medicine, The University of Texas at Austin, Austin, TX
Stadtman Tenure-Track Investigator, Kidney Diseases Section, NIDDK
Associate Professor, W. Harry Feinstone Dept. of Molecular Microbiology & Immunology, Johns Hopkins University, Baltimore, MD
Assistant Professor of Pediatrics, Children's Hospital of Philadelphia, Philadelphia, PA
Associate Professor, Dept. of Pathology and Immunology, Baylor College of Medicine, Houston, TX
Vice President, Medtronic; Associate Professor, Johns Hopkins University, Baltimore, MD
Rheumatologist, Ventura, CA
Medical Oncologist, Cleveland Clinic, Cleveland, OH
Director, Gynecologic Oncology Laboratory, Brigham and Women's Hospital; Assistant Professor of Obstetrics, Gynecology and Reproductive Biology, Harvard Medical School, Boston, MA
Director, Division of Biotechnology Review and Research II, FDA
Director, Office of Science and Technology, NIAMS; Investigator, Translational Immunology Section, NIAMS
Division Chief, Pediatric Rheumatology, Associate Professor of Pediatrics, Indiana University School of Medicine, Indianapolis, IN
Patent Examiner, U.S. Patent and Trademark Office
Postdoctoral Fellow, NIAMS
Senior Scientist, Boehringer Ingelheim, New York City, NY
Rheumatologist, Denver, CO
CSO/COO, ImmPACT-Bio USA Inc. Camarillo, CA
Associate Professor, Dept. of Medicine; Vice Director, Center for Autoinflammatory Diseases, Renaissance School of Medicine, Stony Brook University, Stony Brook, NY
OB-GYN, Cleveland Clinic, Cleveland, OH
Professor of Surgery, Director of Vascular Surgery, University of Maryland, Baltimore, MD
Research/Clinical Fellow, Brigham and Women's Hospital; Assistant Professor of Medicine, Harvard Medical School, Boston, MA
Medical Director, Inpatient Rehabilitation Unit; Clinical Assistant Professor, Rehabilitation Medicine, University of Washington Medical Center, Seattle, WA
Deputy Scientific Director, Acting Chief, Laboratory of Cancer Immunometabolism, NCI
Professor, Dept. of Pathology, University of California San Francisco, San Francisco, CA
Executive Director of Clinical Development, Horizon, Gaithersburg, MD
Program Director, Scleroderma, Fibrosis, and Autoinflammatory Disease Program, NIAMS
Assistant Professor, Dept. of Immunology and Pediatrics, University of Pittsburgh, Pittsburgh, PA
University of Rochester
Investigator, NEI
Assistant Clinical Investigator, NIAID
Program Officer, NIAID
Medical Scientist Training Program, Washington University, Resident in Pathology, Stanford University, Stanford, CA
Investigator, Arboviral Diseases Section, CDC
Postdoctoral Research Fellow, Dept. of Pathology, University of Iowa, Iowa City, IA
Principal Scientist, MilliporeSigma, Rockville, MD
Director, Rapid Response Team, Chan-Zuckerberg Biohub, San Francisco, CA
Spinal Surgeon, UM St. Joseph Medical Center, Towson, MD
Scientist, HiFIBiO Therapeutics, Cambridge, MA
Professor, Pediatrics, Seattle Children’s Research Institute, Adjunct Professor, Immunology, University of Washington, Seattle, WA
Associate Professor of Genetics, Penn Institute of Immunology, University of Pennsylvania, Philadelphia, PA
Assistant Professor, University of Miami, Miami, FL
Associate Professor, Dept. of Infectious Disease, University of Georgia, Athens, GA
Resident, University of Rochester Medical Center, Rochester, NY
Assistant Professor, University of Texas Southwestern Medical Center, Dallas, TX
Physician Scientist, FDA
International
Assistant Professor, University of Bourgogne, Dijon, France
Professor, Director of Rheumatology, University of Dresden, Dresden, Germany
Senior Lecturer, Consultant Hematopathologist, Newcastle University, Newcastle upon Tyne, UK
Associate Professor, Division of Rheumatology, Medical University of Vienna, Austria
Associate Professor of Medicine, Head Physician, Division of Immunology and Allergy; Director, Vaccine and Immunotherapy Center, University Hospital of Lausanne, Lausanne, Switzerland
Associate Professor, Faculty of Biochemistry and Molecular Medicine, University of Oulu, Oulu, Finland
Teaching Fellow, Faculty of Medicine, Imperial College London, London, UK
Professor, Dept. of Gastroenterology, Braine-l'Alleud - Waterloo Hospital, Waterloo, Belgium
Research Director, Head, Laboratory of Integrative Cancer Immunology, INSERM, Paris, France
Director, Dept. of Dermatology, Venerology and Allergology, Charité Medical University of Berlin, Berlin, Germany
Associate Professor, Dept. of Immunology, Chiba University, Chiba, Japan
Dept. of Pediatrics, University of Dresden, Dresden, Germany
Assistant Professor of Rheumatology, University of Occupational and Environmental Health, Fukuoka, Japan
Specialist, Dept. of Oncology, Waikato Hospital, Hamilton, New Zealand
Blood and Marrow Transplant Consultant, University College London, London, UK
Head, Mucosal Immunology Laboratory, Olivia Newton-John Cancer Research Institute, Heidelberg, Australia
Associate Professor, Dept. of Internal Medicine, Keio University, Tokyo, Japan
Professor and Chair, Dept. of Rheumatology and Clinical Immunology, Kyoto University, Kyoto, Japan
Assistant Professor, The First Department of Internal Medicine, University of Occupational and Environmental Health, Fukuoka, Japan.
Professor, Faculty of Medicine and Health Technology, University of Tampere, Tampere, Finland
Associate Professor, Sapienza University of Rome, Rome, Italy
Associate Professor, Dept. of Dermatology, Keio University, Tokyo, Japan
Director, Dept. of Rheumatology, Hamamatsu Medical Center, Shizuoka, Japan
Researcher, Italian National Research Council, Rome, Italy
Scientific Founder, Smilebiotek; Professor, Zhongshan Ophthalmic Center, Sun Yat-Sen University, Guangzhou, China
Professor and Chair, Dept. of Rheumatology and Infectious Disease, Kitasato University, Tokyo, Japan
Professor of Immunology, Tongji Medical University, Huazhong University of Science and Technology, Wuhan, China
Image & Media Gallery






Scientific Publications
Selected Recent Publications
News & Highlights
NIH scientists find treatment for rare genetic skin disorder


NIAMS’ John O’Shea Awarded 2021 Harrington Prize for Innovation in Medicine