
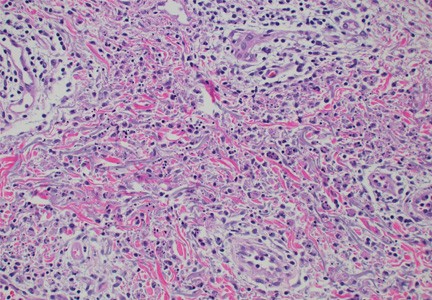
Skin findings in a patient with VEXAS syndrome viewed under a microscope.
Scientists at NIAMS and other institutions have shed light on patterns of skin characteristics seen in patients with VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic) syndrome, an often-deadly inflammatory condition caused by mutations in the UBA1 gene of blood cells. In addition, distinct skin symptoms were linked to specific gene variants in people with the disease. The findings were recently published in JAMA Dermatology.
VEXAS syndrome was first reported by a team of NIH researchers in 2020. People with the disorder often have a wide range of inflammatory symptoms that affect multiple organs. The mutations in VEXAS syndrome happen spontaneously later in life and are not passed from biological parents to their children. The disease tends to occur in the fifth decade or later, and mostly among men, but it can be difficult to diagnose.
"Symptoms of the syndrome can vary and tend to mimic other inflammatory conditions,” said paper co-senior author Edward Cowen, M.D., head of the Dermatology Consultation Service at NIAMS. Cowen also noted that health care providers may not immediately suspect a genetic disease in an older person. Early diagnosis and treatment, however, can help improve patient outcomes and quality of life.
“We saw skin changes in more than 80% of patients, mostly early in the course of the disease,” said first author Isabella Tan, an M.D. candidate at Rutgers Robert Wood Johnson Medical School in Brunswick, New Jersey. Tan was a student in the Vasculitis Translational Research Program under the direction of program chief and co-senior author Peter Grayson, M.D., at the time that the research was conducted.

The team analyzed tissue samples from 60 of 112 study participants who were referred to the NIH Clinical Center in Bethesda, Maryland for suspected VEXAS syndrome. The most reported findings included inflammation of the small blood vessels of the skin, known as leukocytoclastic vasculitis; fever and a painful rash known as neutrophilic dermatosis and inflammation of the skin around the blood vessels, known as perivascular dermatitis.
Most of the skin biopsies revealed a distinct pattern called histiocytoid dermal neutrophilic inflammation, which is sometimes seen in patients with Sweet syndrome, an inflammatory skin disease. This pattern, when combined with leukocytoclasia, may prove useful to a potential diagnosis of VEXAS, according to the researchers.
The team was also able to correlate different skin presentations with the different amino acid changes of the UBA1 gene. “This linking of genotype with phenotype may help us identify distinct patterns of disease, which may facilitate a quicker diagnosis and help inform overall prognosis,” said Cowen.
In the current study, many participants experienced delays in getting an accurate diagnosis before coming to NIH, despite having seen multiple physicians. The researchers are hopeful that as greater attention is brought to VEXAS, people with milder forms of the disease will be more easily identified and diagnosed.
“VEXAS may be newly discovered, but we are seeing that it is not exceedingly rare," said Grayson. “This study should help guide clinicians in recognizing some cardinal features of the disease in skin.”
Tan IJ, Ferrada MA, Ahmad S, et al. Skin Manifestations of VEXAS Syndrome and Associated Genotypes. JAMA Dermatology DOI:10.1001/jamadermatol.2024.1657